今年4月,IQVIA人类数据科学研究所发布了一份名为“研发领域不断变化的格局”的深度研究报告。详细分析了医疗卫生行业的创新和临床发展。药明康德的微信团队也与读者分享了这份报告的亮点。
近日,知名投资人、Atlas Venture合伙人布鲁斯布斯(Bruce Booth)博士在其博客LifeSciVC上发表了他对这份报告中关于临床试验的一些数据的进一步分析。医学领域的临床试验活动在过去几年中增长了35%,2018年开展了超过4700项临床试验。在此背景下,Bruce Booth博士分析了2018年获批新药临床试验的特点,并分享了自己的见解。

2018年获批新药临床试验的特征
随机对照试验(RCT)仍然是批准的黄金标准:近90%的批准的药物监管申请包含RCT。
涉及主动对照组的临床试验越来越普遍。2018年获批的药物中,近一半(46%)在临床试验中纳入了主动对照组。几年前,这一数值仅为20%,这表明在已经存在标准疗法的疾病领域,人们更加注重比较不同疗法的有效性。
在2018年批准的新药中,超过40%(25/59)仅基于一项注册临床试验获得批准。过去“需要两个有适当对照组的3期临床试验”的审批标准在2018年不适用。
12%(7/59)的新药只通过了1期或2期临床试验,没有进行3期临床试验。这些都是特例。
患者暴露(patient exposure)水平和临床开发时间分析
2018年批准的新药,临床试验招募的患者数量差异很大。大约一半的批准要求少于500名患者接受治疗。
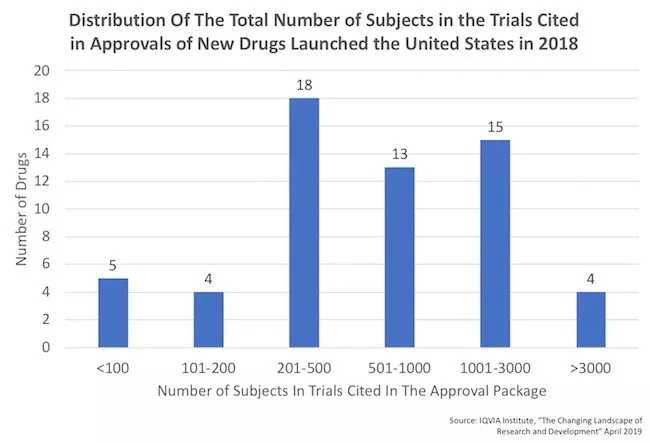
批准的新药申请中临床试验患者分布(来源:参考文献[1])
只有不到100名患者的临床试验数据被纳入五种新药的应用中。这些新药是孤儿药。其中4个新药只需要1个临床试验即可获批,第5个是用于治疗腺苷脱氨酶重度联合免疫缺陷(ADA-SCID)的elapegademase。它是根据两个注册临床试验的结果批准的,共有10名患者。
2016-2018年期间,支持新药上市的临床试验中的患者年暴露指数在1800至3800之间。患者年暴露指数是通过将接受治疗的患者人数乘以接受治疗的年数获得的。是了解药物安全性和远期疗效的重要指标。
孤儿药获得批准平均需要1000个病人年。这个数值虽然比非孤儿药小,但还是高于人们的预期值。
达到这些患者的暴露水平需要大量的研发时间,导致近年来新药的平均临床研发时间仍然在12年以上,没有明显的改善。
尽管纳入孤儿药注册试验的患者数量比非孤儿药少得多(平均430对2,300),但这些R&D项目仍然需要很长时间才能完成。事实上,基于过去四年的平均值,孤儿药的临床试验长度比非孤儿药长12%(7.6年比6.7年)。这是一个惊人的发现。这可能是因为除了罕见的单基因遗传病外,孤儿药的识别还包括许多其他类型的适应症。而且孤儿药研发项目需要在更小数量的患者身上积累足够的安全性和疗效数据。
临床开发需要注意的问题
Bruce Booth博士将这些数据与他自己作为早期投资者的经历结合起来,分享了以下见解:
在随机对照试验中使用主动对照将变得更加普遍,即使是对孤儿药。在研发过程中尽早了解在研治疗和现有标准治疗的区别变得越来越重要。试图不使用主动控制来节省临床试验成本的策略是不可取的。设计正确的临床试验,说服投资者支持这些试验,不要偷工减料。
从历史经验来看,单臂测试和没有适当控制的测试通常在后来面临更多的问题。比如在癌症研究中,这往往会导致假阳性结果,导致后期临床研发失败。一些罕见疾病甚至缺乏相关长期自然疾病史作为对照。因此,在这种情况下,药物开发公司应在临床前研究阶段投入早期观察性临床研究,旨在积累足够的疾病历史对照和比较疗效的基线指标。
对于“主流”孤儿药,它们未来仍需要约1000个病人年的数据才能获得监管机构的批准。对于非孤儿药,需要的数据量会在2000~3000左右。具有显著疗效的基因和细胞疗法可能需要较低水平的患者暴露。这些一次性治疗受益于通过一次治疗后的随访持续积累的患者年数据。
尽管存在特殊的成功案例,但很少有进展中的疗法可以直接从1期或2期临床试验中获得批准,除非它们在早期试验中显示出非常显著的疗效。药物R&D团队不能依赖这种加速审批策略作为药物研发的基本方案
参考资料:
[1]让新药上市具有临床意义。检索于2019年5月17日,来自。
[2]不断变化的研发格局。检索于2019年5月17日,来自
关注药明康德微信官方账号
