柯文智康
美国FDA的现场检查是根据FDA检查员对被检查企业在生产过程中是否遵守cGMP要求的要求,同时检查企业的所有申请文件是否与现场文件中记录的数据一致、准确、完整、可靠。FDA将重点对六个职能部门进行全面检查,包括质量保证部门、工厂和设备、原料、生产、包装和实验室。如发现问题,将要求公司以“483表”的形式说明问题;如果没有得到满意的解释,将向企业发出警告信。FDA警告信是向全球企业和个人发出的关于其产品、生产、经营或其他活动违反FDCAct( 《联邦食品、药品和化妆品法案》)的正式公告。
近年来,中国医药产业国际化进程不断加快,越来越多的药品在FDA注册。中国制药企业也受到了FDA的现场检查。为了让医药行业更清楚地了解FDA现场检查的要点和相关数据,笔者对这篇文章进行了翻译和整理,供大家参考。
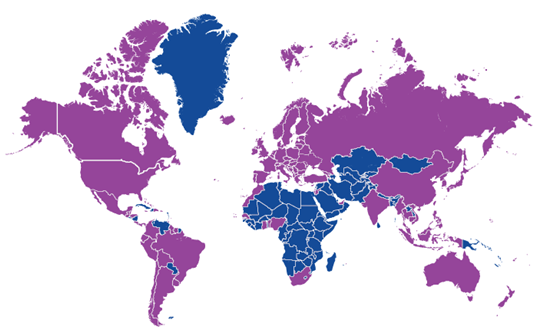
据了解,自1955年以来,FDA一直在进行国际检查,以支持《联邦食品、药品和化妆品法案》。1976年,根据《联邦食品、药品和化妆品法案》医疗器械修正案,FDA也将国外医疗器械和诊断制造商纳入国际检验计划。世界地图中的紫色区域是最新的FDA法规事务办公室(ORA)检查的国家和地区。ORA的作用是最大限度地提高FDA监管产品的合规性,并最大限度地降低与这些产品相关的风险。
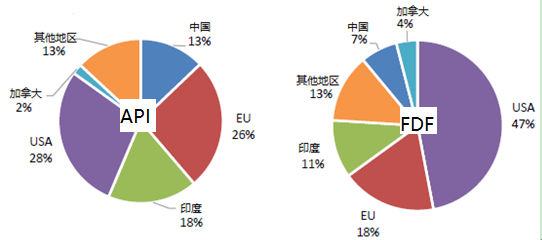
截至2019年8月,美国市场72%的原料药生产厂和53%的FDF生产厂位于美国境外,如下图所示。
在2012年通过《食品和药品管理局安全与创新法》 (FDASIA)之前,法律要求FDA每两年对美国的生产设施进行检查,但对国外设施的检查频率没有类似的授权。2012年,FDA修订了《联邦食品、药品和化妆品法案》的第510(h)节,将对生产场所的监督检查频率从原来的固定的至少每两年一次改为基于风险的检查计划,以制定策略,将是否存在对患者造成伤害的最大风险作为检查计划中的优先因素,并使用有限的检查资源检查高风险的生产场所,以确保所有生产场所生产的所有药物(包括原研药和仿制药、处方药和非处方药)
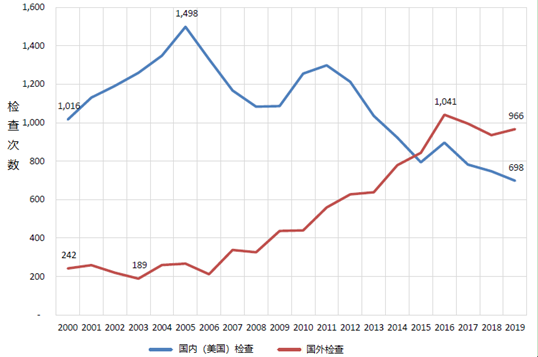
自2015年以来,CDER对外国企业的检查多于美国国内企业,如下图所示。
自2005年起,FDA开始在CGMP日常监督检查中实施基于风险的选址模型(SSM),对生产场所进行排序,并制定检查计划,即现场监督检查清单(SSIL)。
根据CDER的检查清单,截至2016年7月,有965家外国工厂从未接受过FDA的检查。到2019年,其中495家(51%)之前接受过FDA的检查,另有359家工厂(37%)被从检查名单中删除,因为它们不再是FDA检查工作的一部分,原因有很多:例如,它们已经倒闭,不再为美国服务。此外,52家(6%)工厂拒绝检查;37家(4%)工厂因为到不了(比如旅游警告)而无法检查;两个(2%)工厂没有药品运输。
SSM是FDA监督检查优先程序的核心,是目前FDA检查国内外生产设施的统一方法。这项工作由药品质量办公室(OPQ)下属的监督办公室进行。FDA使用该模型根据风险因素计算检查表中每个设施的得分。SSM的风险因素包括:
固有产品风险:根据剂型、给药途径或无菌的特点,不同类型的产品有不同程度的风险。例如,生产无菌注射剂的工厂将比生产口服胶囊的工厂具有更高的固有产品风险。
设施类型:风险等级可能因工厂执行的操作而异。生产毒品的工厂比只包装毒品的工厂风险更大。
患者暴露:生产的产品的数量和类型越多,患者接触本职能领域生产的产品的可能性就越大
检查历史记录:在以前的检查中不符合既定质量标准的设施被认为比过去符合标准的设施具有更高的风险。
010-5900随着距离上次设施检查时间的增加,不符合既定质量标准的风险增加,重新检查的必要性也增加。
上次检查后的时间:与很少或没有重大危险信号的设施相比,产品召回或制造商或患者报告与设施相关的质量问题将增加风险得分。
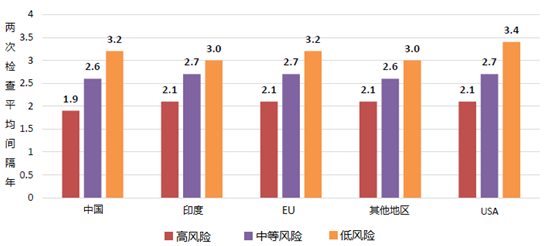
将一个设施的得分与FDA检查清单中的其他设施的得分进行比较,并根据风险对它们进行排名,而不考虑位置。如果考虑设施的三个相对静态因素(固有产品风险、设施类型和患者暴露),高风险设施的检查间隔中位数为2.1年,从2011年12月到2019年6月。无论在什么位置,所有高风险设施的检查频率几乎相同。详见下图。
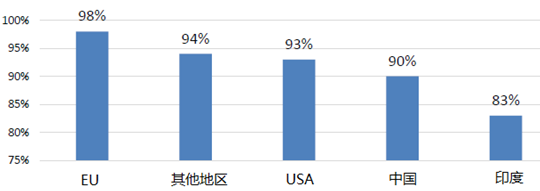
世界上大部分人用药品生产设施的最终检查结果是可接受的(90%的结果是NAI不需要整改或VAI需要自愿整改)。但印度的设施可接受检查结果低于其他国家和地区。中国的检验结果好于印度,但低于欧盟、美国等地区。详细数据见下图,截至2019年8月。
根据CGMP,外国和国内药品制造商必须满足相同的监管要求。如果工厂在检查中没有达到CGMP标准,FDA有一系列的监管工具,可以用来鼓励公司修改和改进其生产过程,以达到合规性。这些监管工具包括警告信、进口警告、禁令和没收。如果FDA在后续检查中发现这些设施仍然不符合CGMP标准,它可能会酌情将问题升级。
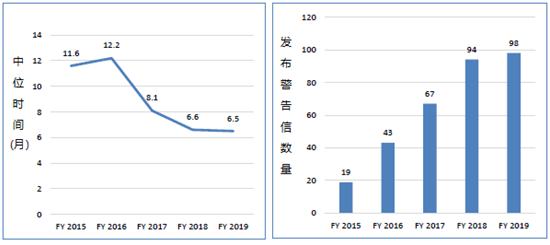
从2015年到2019年,从检查结束到发出警告函的整体平均时间改善了44%,从超过11个月缩短到6.5个月。同时,警告信的数量从19封增加到98封。
危险信号:
进行国外检查的FDA人员分类如下:
(1)在美国进行国内和国外检查的检查员;
(2)在美国专门从事外国检查的检查员;
(三)海外分支机构检查员。
大多数国外检查是由前两类检查员进行的。