监管环境的明显改善和跨国公司报告策略的改变普遍缩短了获得上市许可所需的时间,六个监管机构之间的协调也有所改善。在过去的10年里(2009-2018年),批准上市的新药数量也有所增加。这六个监管机构包括欧洲药品管理局(EMA)、美国美国食品药品监督管理局(FDA)、日本药品和医疗器械管理局(PMDA)和加拿大卫生部。
监管科学创新国际研究中心(CIRS)在《R&D简报》第70期发表了一项名为《2009-2018年6大监管机构批准的新药:关注加速审评通道和孤儿药状态》的新研究,该研究统计了新活性物质(NAS)的批准情况,并发现了以下发现。
这六个监管机构包括欧洲药品管理局(EMA)、美国美国食品药品监督管理局(FDA)、日本药品和医疗器械管理局(PMDA)、加拿大卫生部、瑞士医疗和澳大利亚药品管理局(TGA)(如图1所示)。
CIRS的统计数据显示,六个监管机构批准的品种数量从2009-2013年的16个NAS增加到2014-2018年的52个NAS,这表明在此期间有更多的产品实现了国际化。
影响一种新药提交监管部门审批的总时间的潜在因素包括公司的战略、审评程序的实施和类型、产品类型及其治疗领域。特别是,FRP(加速审查通道)、孤儿药指定和申请人规模是影响提交和批准策略的主要因素。
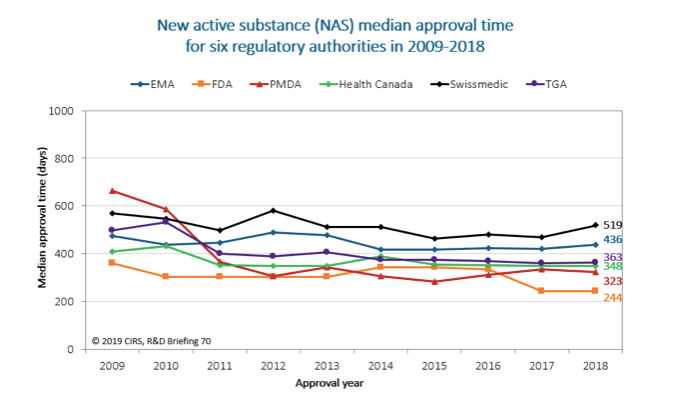
图1:监管环境的改善有助于获得上市许可所需时间的普遍缩短。(审批时间是从提交申请到监管机构批准所需的时间。时间包括监管机构和申请人花费的时间。EMA批准时间包括欧盟委员会花费的时间。)
关注加速审评通道
六个监管机构实施了一个特定的FRP,即加速评估程序(EMA的“加速评估”,Swissmedic的“绿色通道”和其他四个监管机构的“优先评估”),旨在加速有前途的新活性物质的评估过程。TGA从2017年开始实施优先评价制度,2018年首次通过该制度批准新药。一般来说,通过加速审查程序所需的时间比标准程序短。
2018年,FDA(73%)的加速审查比例最高,其次是加拿大卫生部(35%)、PMDA(28%)、Swissmedic(13%)、EMA和TGA(10%)。
在六个监管机构中,FDA提供(或可以使用)最快速的评估渠道(FRP)。对于医疗需求未得到满足的地区,加快药物的评估和/或批准,并改善药物的可及性。2018年,FDA批准的NAS品种中有75%受益于至少一种可用的FRP(不包括孤儿药)。在其他机构中,FRP的受益率介于TGA的10%和加拿大卫生部的41%之间。(参见图2)。
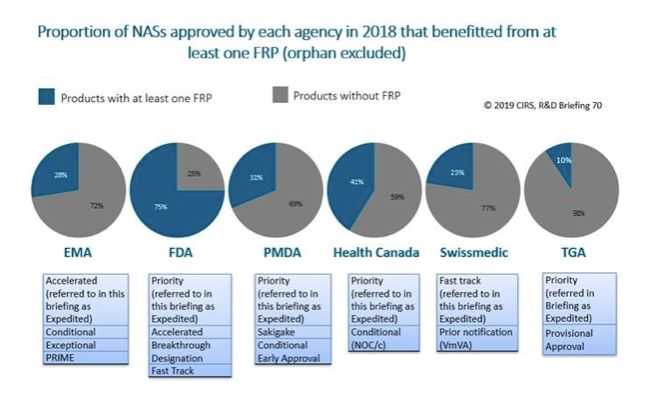
图2:在六个国家的监管机构中,FDA提供(或可以使用)最快速的评估渠道(FRP)。对于医疗需求未得到满足的地区,加快药物的评估和/或批准,并改善药物的可及性。
关注孤儿药状态
被六个监管机构指定为孤儿药的NAS数量从2009-2013年的25%增加到2014-2018年的38%。2018年,FDA批准的孤儿药最多(35个),PMDA批准的孤儿药最少(8个)。加拿大卫生部尚未实施孤儿药物政策;然而,在2018年,该机构批准了15种被FDA,EMA或TGA指定为孤儿药的NAS。
2018年,FDA对孤儿药的中位审批时间最短(243天),因为88%的品种是通过加速审评审批的。在六个监管机构中,只有EMA的孤儿药审批时间中位数超过非孤儿药。2018年Swissmedic的这些时间指标比较接近。2018年TGA批准的孤儿药中,有20%是通过刚刚开始的优先审评程序获得批准的。
在2014-2018年6家监管机构批准的52个NAS品种中,只有10个NAS品种在所有监管机构中被指定为孤儿药,这表明监管机构之间指定孤儿药的标准存在一定差异。一般来说,孤儿药NAS的中位提交时间差比非孤儿药NAS长。
事实上,大多数孤儿药NAS都是由非顶级企业注册的,这凸显了小规模企业在促进创新方面的重要作用。
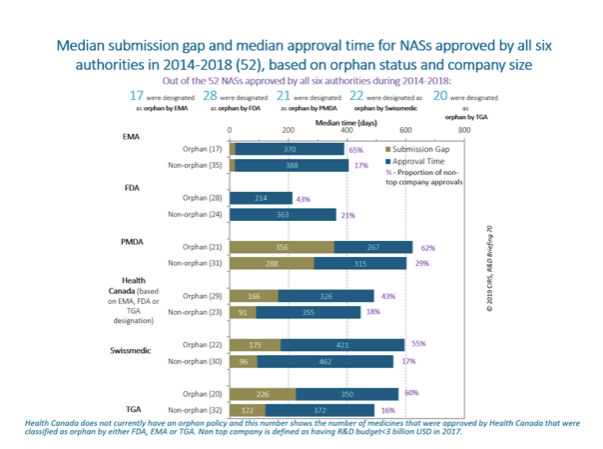
图3: CIRS的统计数据显示,大多数孤儿药非专利药品是由非顶级企业注册的,这凸显了小型企业在促进创新方面发挥的重要作用。
